Bienvenue sur Waldenström France
Les actualités

JOURNÉE MONDIALE MALADIE DE LA WALDENSTRÖM
La fondation américaine IWMF initie pour la première fois la journée mondiale de la macroglobulinémie de Waldenström le 17 avril date anniversaire de la naissance du médecin suédois Jan Waldenström qui a découvert cette maladie….
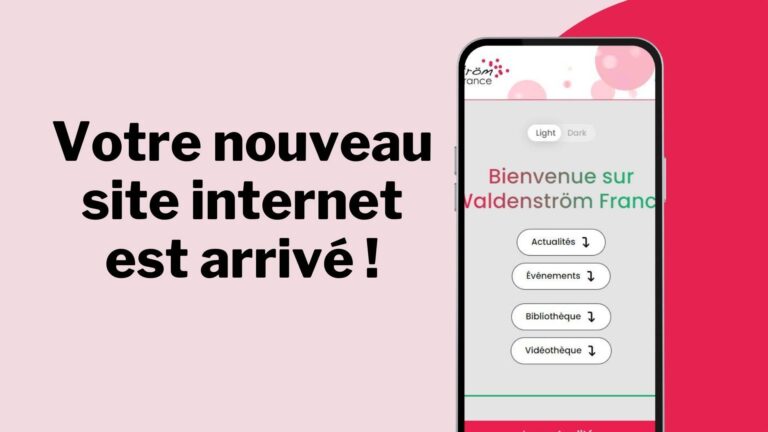
Le site de Waldenström France fait peau neuve
Bienvenue sur votre nouveau site internet ! Afin de faciliter l’accès aux informations pour les personnes en quête de renseignements et de support sur la maladie de Waldenström, notre association met à votre disposition son nouveau…

Bonne année 2024
Au nom du Conseil d’Administration de l’Association Waldenström France, permettez-moi de vous adresser tous mes vœux de très belle et heureuse Année pour vous-même ainsi que pour tous ceux qui vous sont chers. Que 2024…
Événements
Bibliothèque
Fiche d’information sur les jeunes patients atteints de la Macroglobulinémie de Waldenström
Fiche d’information sur les jeunes patients atteints de la Macroglobulinémie de Waldenström Cette Fiche d’information est à l’usage des jeunes patients atteints de la macroglobulinémie de Waldenström Niveau de connaissances :
Guide du patient
Guide du patient – Informations essentielles Ce nouveau livret est un condensé d’informations qui résume en quelques pages toute les réponses aux questions que l’on se pose en découvrant la maladie de Waldenström. Rédigé en 2023 par l’IWMF…
guide à destination du personnel infirmier
Informations essentielles : guide à destination du personnel infirmier Nouveau guide sur la macroglobulinémie de Waldenström, réalisé et traduit par l’IWMF en 2023, à l’attention des infirmiers(ières). Niveau de connaissances :
Guide à destination des médecins
Informations essentielles : guide à destination des médecins Nouveau guide sur la macroglobulinémie de Waldenström, réalisé et traduit par l’IWMF en 2023, à l’attention des médecins. Niveau de connaissances :